Our research activity focuses on the development of theory and algorithms for the calculation of molecular properties and molecular spectra of systems embedded in external complex environments based on the first principles of quantum mechanics.
All current projects may be grouped according to five main research directions:
- Development of Fully Atomistic Embedding Approaches for Computational Spectroscopy
 Our research focuses on the development and implementation of fully polarizable multiscale QM/MM approaches, for the calculation of molecular properties and spectroscopies of systems embedded in external environments. For a recent review see ACS Phys. Chem. Au 2022.
Our research focuses on the development and implementation of fully polarizable multiscale QM/MM approaches, for the calculation of molecular properties and spectroscopies of systems embedded in external environments. For a recent review see ACS Phys. Chem. Au 2022.
Moreover, we have extended these models to treat chiroptical properties. This is particularly relevant since most chiroptical responses are measured for systems interacting with an external environment, which can have huge effects on both the absolute value and sign of the spectral signals. Chiroptical properties are complex phenomena involving the molecular response to both the electric and magnetic components of the radiation field. Thus, being able to model both fields reliably is mandatory to obtain accurate spectra. For more details see J.Phys. Chem. Lett., 2016, 7, 3585; J. Chem. Theory Comput., 2013, 9, 1880.
Development of Fully Atomistic Approaches for Modeling the Optical Response of Plasmonic Substrates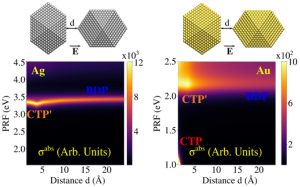 We develop fully atomistic classical models capable of describing the optical properties in the frequency domain of plasmonic substrates such as graphene or metal nanoparticles. Our models prove to be reliable against ab-initio results. Moreover, the affordable computational cost of our models, allows us to apply them to realistic-sized structures, still retaining a full atomistic description of the system.
We develop fully atomistic classical models capable of describing the optical properties in the frequency domain of plasmonic substrates such as graphene or metal nanoparticles. Our models prove to be reliable against ab-initio results. Moreover, the affordable computational cost of our models, allows us to apply them to realistic-sized structures, still retaining a full atomistic description of the system.
For more details see ACS Photonics, 2022, 9, 3025; J. Phys. Chem. Lett., 2020, 11, 7595; Nanoscale, 2019, 11, 6004.- Application of Computational Protocols to Molecular Spectroscopy of Relevant Chemical Systems
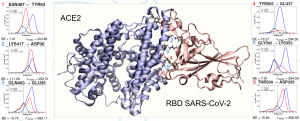 We apply our methods and codes to describe and predict various spectroscopic phenomena, from purely electronic to purely vibrational or vibronic spectra of systems of applicative interest. Using our tools, we have recently studied biologically relevant systems like the ACE2⋅⋅⋅RBD SARS-CoV-2 complex (also with the SARS-CoV-2 variants), the insertion of drugs into model cell membranes, and the intercalation of anticancer drugs into DNA. Some works arise from collaboration with experimental groups. A few examples can be found in ChemBioChem, 2021, 22, 724; J.Phys. Chem. B, 2021, 125, 10383; J. Chem. Theory Comput., 2022, 18, 1765.
We apply our methods and codes to describe and predict various spectroscopic phenomena, from purely electronic to purely vibrational or vibronic spectra of systems of applicative interest. Using our tools, we have recently studied biologically relevant systems like the ACE2⋅⋅⋅RBD SARS-CoV-2 complex (also with the SARS-CoV-2 variants), the insertion of drugs into model cell membranes, and the intercalation of anticancer drugs into DNA. Some works arise from collaboration with experimental groups. A few examples can be found in ChemBioChem, 2021, 22, 724; J.Phys. Chem. B, 2021, 125, 10383; J. Chem. Theory Comput., 2022, 18, 1765. - QM/Atomistic Frameworks for Surface Enhanced Molecular Properties
 We are currently working on the extension of our frequency-dependent atomistic models within a QM/classical formalism, for the study of the enhanced response properties of molecules perturbed by plasmonic structures in their vicinity. These computationally affordable, yet atomistic, models constitute a cornerstone for the study of phenomena such as Tip/Surface-Enhanced Raman (TERS/SERS) for the in-silico design of biosensors, among others. See J. Chem. Theory Comput. 2023, 19, 12, 3616.
We are currently working on the extension of our frequency-dependent atomistic models within a QM/classical formalism, for the study of the enhanced response properties of molecules perturbed by plasmonic structures in their vicinity. These computationally affordable, yet atomistic, models constitute a cornerstone for the study of phenomena such as Tip/Surface-Enhanced Raman (TERS/SERS) for the in-silico design of biosensors, among others. See J. Chem. Theory Comput. 2023, 19, 12, 3616.